我校贾传义教授在国际顶级期刊JACS发表重要学术成果
近日,贵州师范学院贵州省纳米材料模拟与计算重点实验室贾传义教授(特聘)与中国科学技术大学江俊教授合作,在双原子复合材料研究方向取得突破性进展,相关成果发表在顶级化学期刊Journal of the American Chemical Society(化学类一区,IF:14.5)上。贵州师范学院为论文的第一署名单位,贾教授为该论文第一作者,李波老师为共同第一作者。此工作对我校化学类学科建设与相关科研工作的开展具有很好的推动作用。

研究背景与学术意义
过渡金属双原子复合催化剂因具有独特的电子特性、均匀的活性位点以及极高的原子利用率,已成为能源、环境催化领域(尤其是CO2还原方向)的新热点。然而,高度分散的金属原子存在易团聚失活的天然缺陷。此外,因复合体系表面结构复杂、反应路径多样,这一前沿方向还存在着微观增效机制和构效关系不清晰等问题。以上难题的解决可以极大加快相关高效催化剂的合成、筛选和应用进程,这对我国“双碳”战略目标的实现也具有非常重要的现实意义。
针对上述挑战,本文以C2N负载的过渡金属双原子为基本模型,对其表面CO2的电、热还原过程进行了系统的理论研究(如图1所示)。结果表明通过双金属的种类调节可以显著改变催化性能,其中Co-Ni因具有较高的活性和选择性,表现最佳。后续的定量构效关系探索发现:对于反应过程中的基元步骤,独立的描述符参数(如:关键吸附物、电荷转移、光谱结构等)就可以准确的反应其稳定性和活性;但对于整体反应过程,只有用基于SISSO拟合的综合描述符才能进行精准预测。以上成果揭示了不同描述符参数在CO2还原过程中作用机制和应用范围,从而为相关高效催化剂的理性设计与定向合成提供了科学的理论指导。
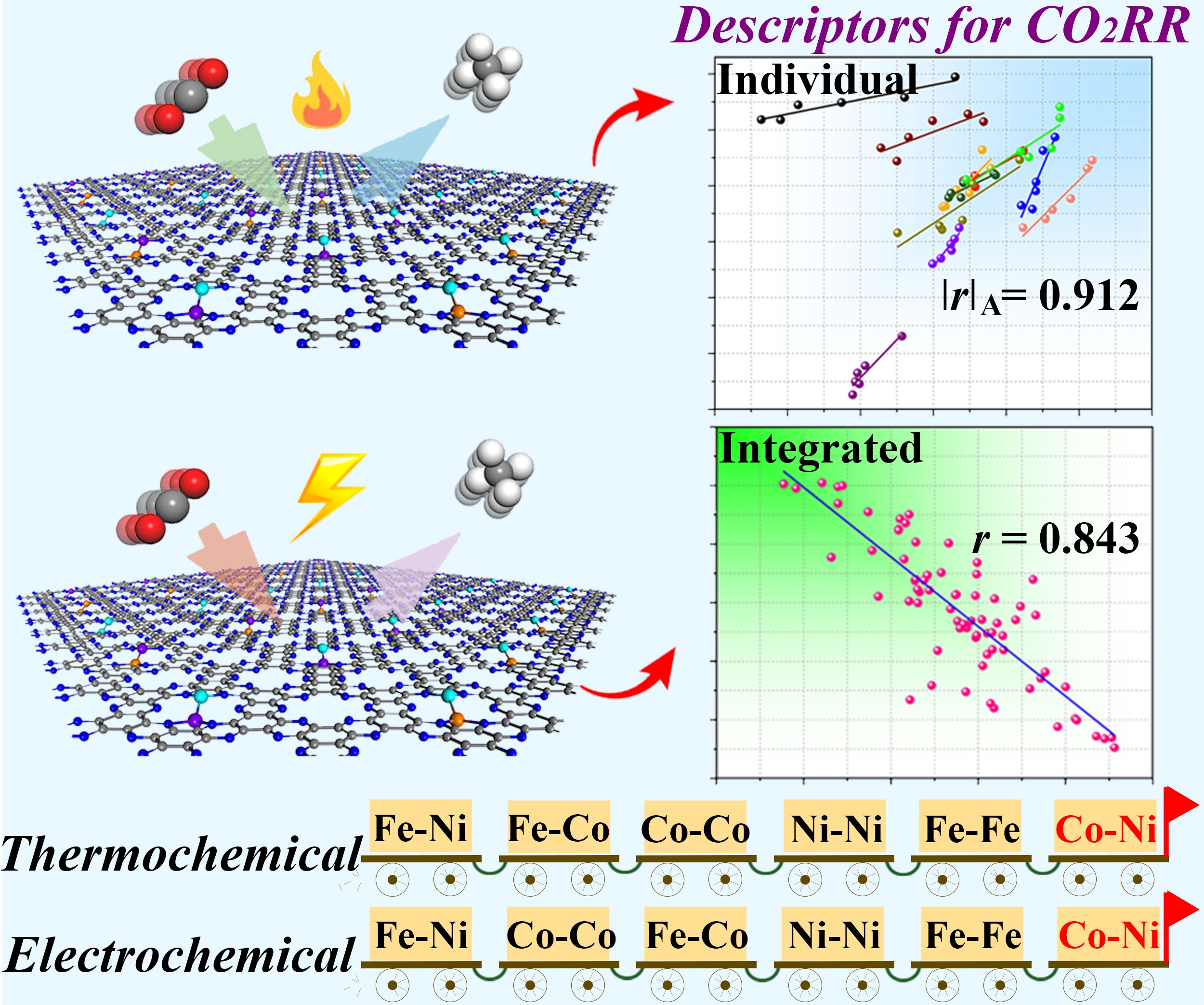
图1. CO2电/热还原模型与独立/综合描述符线性关系示意图。
主要研究内容
为了确保研究成果的可靠性,作者首先对不同模型的稳定性进行了详细的热力学和动力学测试。计算结果表明各种双金属组合在C2N表面的迁移能垒均大于2.0 eV,且能够在500 K下长时间保持稳定(如图2所示)。可见,二维C2N载体对双原子催化剂具有极强的稳定作用。
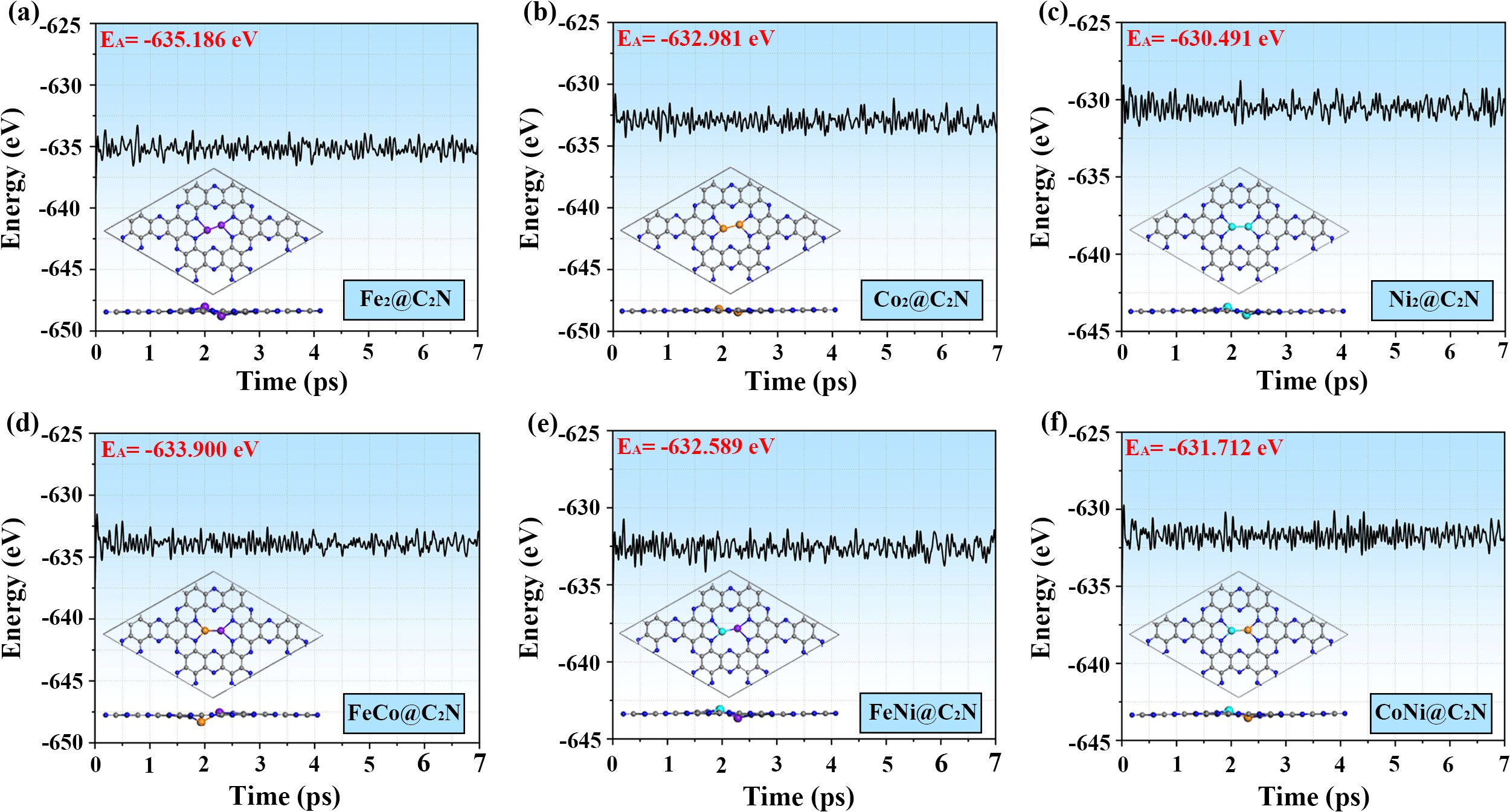
图2. 双金属动力学稳定性测试图。
基于上述模型,作者系统研究了Fe-Fe、Co-Co、Ni-Ni、Fe-Co、Fe-Ni、Co-Ni等多种双金属表面的催化过程(如图3所示)。经过对不同催化剂表面反应机理的比较发现,Fe-Fe、Ni-Ni、Fe-Co、Fe-Ni金属组合更倾向于羧基机理(Carboxyl route),而Co-Co、Co-Ni表面更倾向于甲酸机理(Formate route)。其电催化活性顺序为:Co-Ni (UL = -0.525 V) > Fe-Fe (UL = -0.582 V) > Ni-Ni (UL = -0.610 V) > Fe-Co (UL = -0.710 V) > Co-Co (UL = -0.873 V) > Fe-Ni (UL = -1.013 V)。此外,为了验证理论模拟结果的可行性,作者还用“双参比”方法进行了溶剂化和恒电势验证,从而使理论结果能更好的贴近实际实验过程。
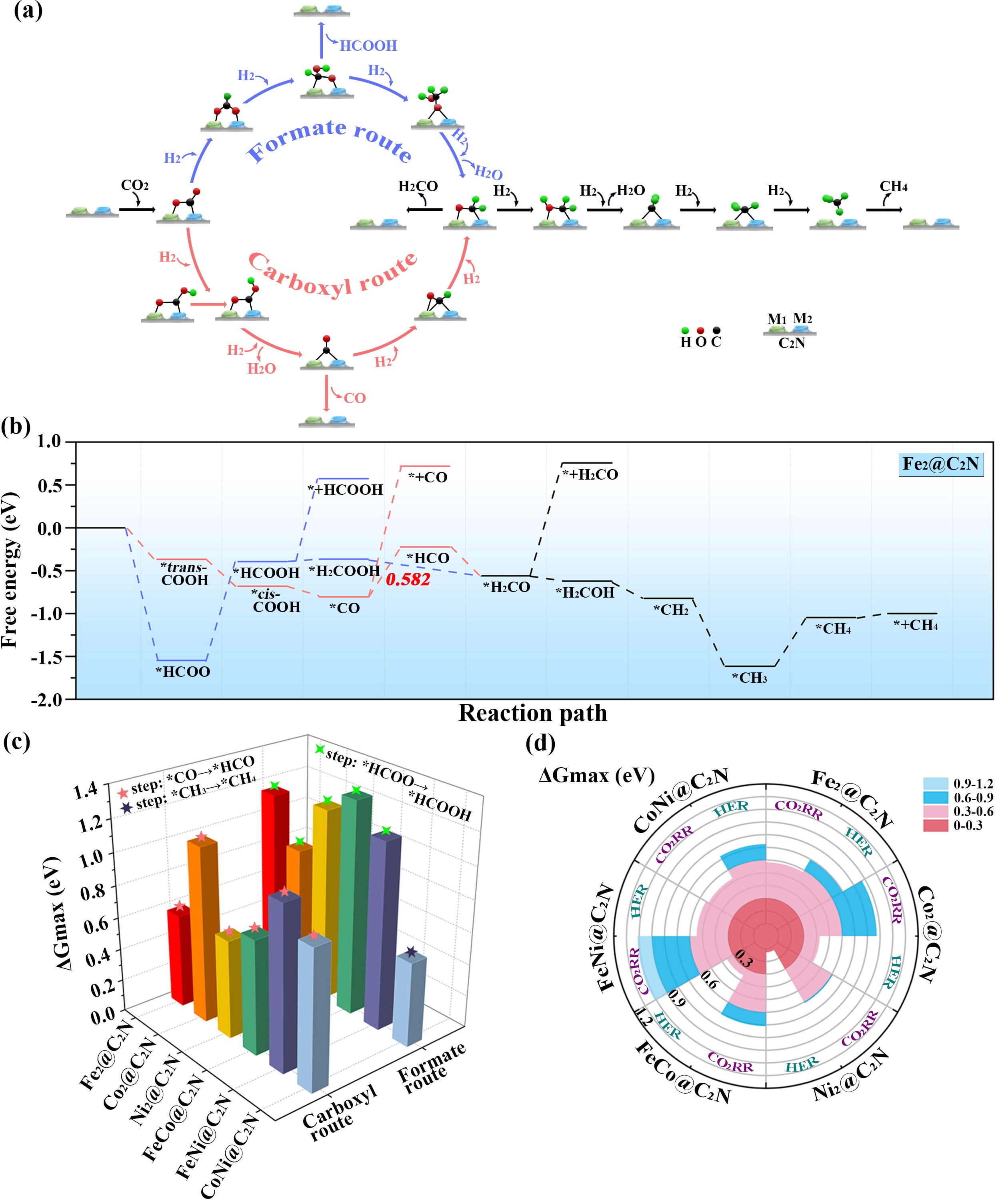
图3. CO2电还原机理和活性对比图。
在大量构效关系分析基础上,作者进一步对各类描述符参数进行了系统性探索。关键吸附物描述符的研究表明:采用CO2、CO等简单分子可以准确预测多种复杂中间体的吸附稳定性,但其普适性和精度较低。电子结构描述符的研究表明:电荷转移、金属带电量等描述符可以实现所有基元反应中间体吸附稳定性的精准预测;而对于反应活性,d轨道中心变化、电荷差、平均键长等描述符表现最佳(如图4所示)。
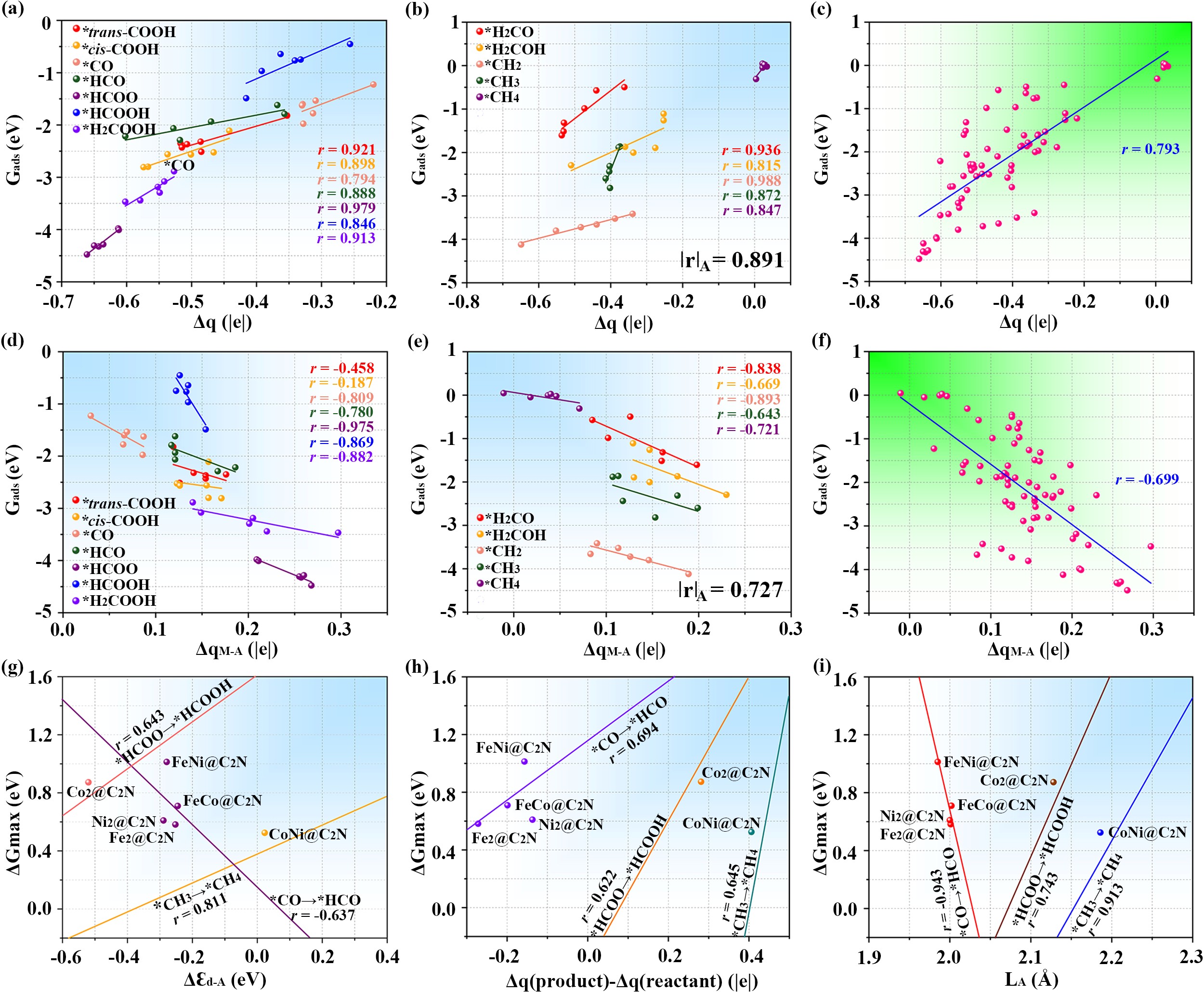
图4. CO2电还原性能与不同描述符参数的线性关系图。
接下来,本工作又详细探究了CO2的热还原过程。如图5所示,不同金属组合最优热催化机理分别为:Fe-Fe、Co-Co、Ni-Ni、Fe-Ni、Co-Ni表面羧基机理;Fe-Co表面甲酸机理。能垒顺序为:Co-Ni (Ea = 0.871 eV) < Fe-Fe (Ea = 0.952 eV) < Ni-Ni (Ea = 0.994 eV) < Co-Co (Ea = 1.106 eV) < Fe-Co (Ea = 1.269 eV) < Fe-Ni (Ea = 1.300 eV)。显然,具有最低能垒的Co-Ni活性最高。进一步的构效关系探索表明,对于热反应活性来说,反应物吸附稳定性、电荷转移、金属带电量等常规描述符与其反应活性都有很好的线性关系。基于光谱结构对比,作者还提出了全新的金属伸缩振动光谱描述符,其较高的精度和较强的实用价值可以为实验上相关催化剂的设计提供新思路。
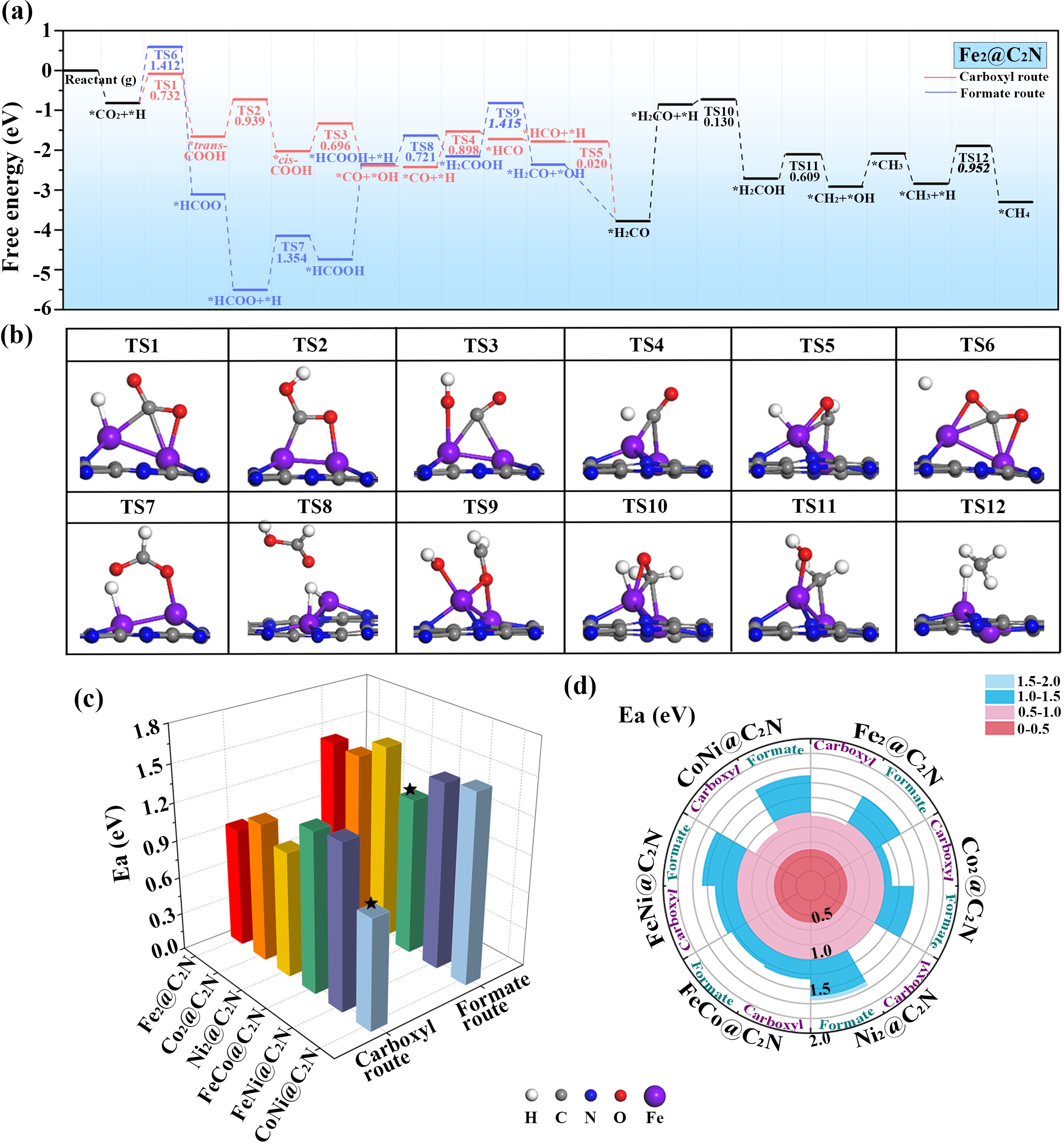
图5. CO2热还原机理和活性对比图。
通过电、热催化过程的对比研究,作者发现虽然独立的描述符参数能很好反应基元步骤中的吸附稳定性和活性,但将其应用于整体活性研究时,精度会明显降低(如图6所示)。为了找出能够准确反应整体活性的高精度描述符,作者采用SISSO分析,拟合了多种综合描述符参数。从图6中可以看出,与独立描述符相比,综合描述符的精度会有大幅度提升。更有趣的是,采用综合描述符可以实现电、热催化过程的统一比较,这极大的简化了分析难度,并显著提升了描述符的普适性和迁移性。
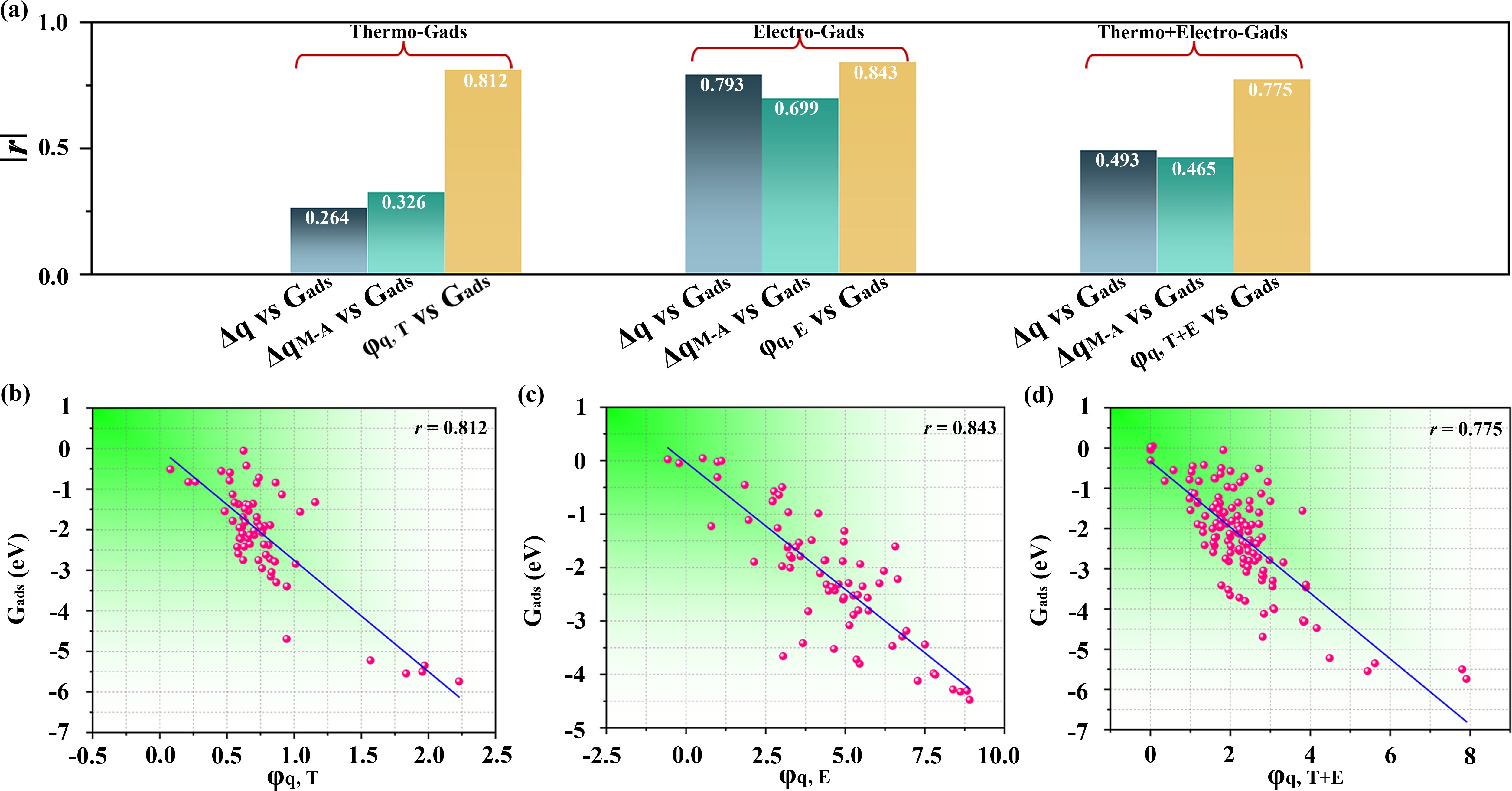
图6. 综合描述符线性关系图。
本文创新与应用前景
基于第一性原理计算,作者对双原子催化剂表面CO2电、热催化机理及其构效关系进行了系统性探索,并提出了关键吸附物、电荷转移、光谱结构等多种高精度的独立描述符参数。此外,基于SISSO分析,作者还提出了能够准确反应整体活性的综合描述符。以上探索结果对理论指导实验定向合成具有重要的参考价值,并为后续采用人工智能技术进行普适性描述符探索提供了新策略。文中构效关系的研究思路可以加速人们对效率高、实际应用性强的双原子复合催化材料的研究步伐,进而大幅提升CO2还原催化剂的筛选和合成效率。
总结与展望
本工作通过对双原子复合催化剂表面CO2电、热还原机理的研究,提出了多种独立和综合描述符参数。结合最新的机器学习(J. Phys. Chem. Lett. 2025, 16, 1424)和理论指导实验的相关研究成果(Angew. Chem. Int. Ed. 2024, 63, e202404418),贾教授团队已初步实现了理论、实验和机器学习的交叉融合。在后续工作中,其团队将继续针对原子级分散的复合催化剂表面CO2还原过程进行更为系统的研究,以期利用学科交叉优势实现催化剂的理性设计,进而筛选出一些催化活性高、稳定性好的新型复合催化材料,并指导实验进行定向合成。
作者简介

贾传义,贵州师范学院贵州省纳米材料模拟与计算重点实验室特聘教授,2014年获山东大学晶体材料国家重点实验室材料学博士学位,同年入职贵州师范学院。主要从事理论化学与人工智能的交叉研究,现聚焦“双碳”领域已对大量高分散复合催化剂开展了系统的构效关系探索,理论计算结合机器学习实现了若干重要催化剂活性和选择性的精准预测,并指导实验进行了定向合成。目前已在Nat. Commun., J. Am. Chem. Soc., Angew. Chem. Int. Ed., Adv. Mater., ACS Catal.等国际著名期刊发表SCI论文近40篇(总引用超1600次);申请到国家自然科学基金项目两项、省级自然科学基金项目两项;2017年入选“西部之光”访问学者、2020年入选贵州省高层次创新型人才“千层次”、2020年获得贵州省自然科学奖三等奖。
该研究成果得益于贵州师范学院在人才引进与人才培养等政策方面的支撑,研究工作得到了贵州省纳米材料模拟与计算重点实验室、贵州省能源与信息材料人才基地、贵州省科学云大数据开发及其产业应用协同创新中心等平台的支持。

