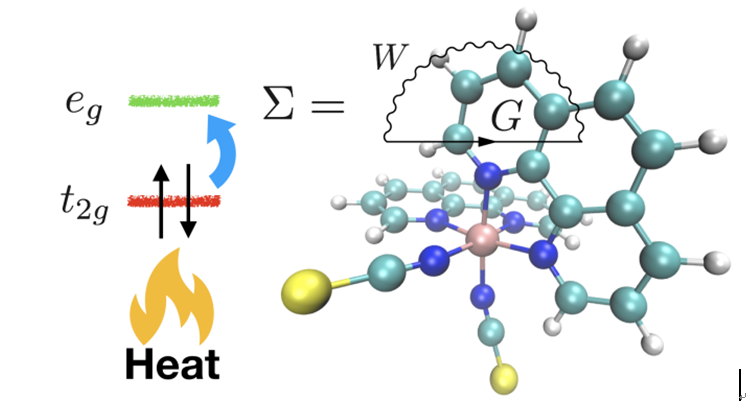
在特定的配位场中,Fe(II)化合物可以在外界的温度变化、压力和光照刺激下实现低自旋(S=0)与高自旋(S=2)状态之间的转换。这种可逆的自旋转变行为(自旋交叉)广泛存在于自然界,并参与生命体以及地球内部重要的化学物理过程。同时,由于分子层次上的自旋双稳态特性,这种材料有望用于制造分子基自旋电子器件,如存储器、开关、传感器等。针对这类体系的基础研究目前主要集中于理解热致自旋交叉现象。这是一个典型的熵驱动过程:随着温度的升高,自旋和振动自由度贡献的熵引起的自由能降低足以克服电子自由能升高时,自旋跃迁就会发生。在某一温度下,高自旋与低自旋分子数将达到平衡,这个温度就是所谓的临界温度。几十年来,通过计算机模拟临界温度来辅助设计自旋交叉材料一直是计算材料领域的一个重要目标。
不幸的是,这些化合物含有强关联3d电子,从而导致不同自旋态之间微妙的能量平衡很难用普通的理论方法再现:基于波函数的方法除了简化的自旋交叉模型外,模拟真实材料体系需要消耗巨大的计算资源;另一方面,密度泛函理论虽然在计算量上可以承受,但其精度不足以描述好自旋转变过程。由于准确性与计算成本之间的平衡,Hubbard U校正的局域密度近似(LDA+U)是一个非常有希望的工具。尽管LDA+U方法在绝热自旋能级差的计算上取得了巨大成功,但众所周知,U的取值依赖于对实验数据的拟合,这很大程度上使得LDA+U的预测能力大打折扣。另一方面,静态的U值不能很好地考虑不同材料电子结构的差异。
最近,贵州省纳米材料模拟与计算重点实验室的研究人员提出了一种通过第一性原理计算获取U值的方法。这个计算框架主要基于解决多体问题的准粒子GW近似。GW方法以屏蔽库仑相互作用的形式有效地描述了电子关联效应;该近似基于严格的代数推导,是一个无参数理论,目前已成功应用于分子和固体的电子结构与激发态性质的计算。本研究利用GW方法获得晶体场分裂能,接着除去自旋态分裂能的贡献得到金属中心3d电子的平均成对能,即近似的U值。通过对一系列具有不同配位场的化合物进行测试,发现用这种方法确定的U值是依赖于局部化学环境的,由此得到的LDA+U方法可以再现所有化合物的自旋基态,并且在临界温度计算上明显克服了静态U的缺点,与实验结果符合较好。
随着计算能力的提高,将这种计算方法推广到分子固体材料是很有希望并且必要的。因为分子间的相互作用和协同效应对自旋交叉材料的性能有非常重要的影响。对于分子晶体,取得依赖于化学环境的U值将使这个领域的研究向前迈出一大步。本研究所提出的计算方法为自旋交叉材料的合理设计提供了一条有前途的新路径。
这项研究已于2019年10月1日在《The Journal of Chemical Physics》期刊在线发表
(https://aip.scitation.org/doi/full/10.1063/1.5124239),通讯作者张亚超。本研究受到国家自然科学基金资助,理论计算受到贵州省凝聚态材料与大分子模拟高性能计算平台支持。